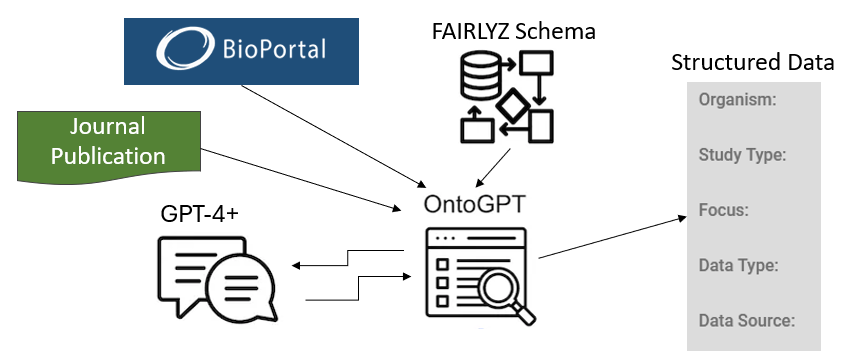
In Part 1, we introduced a brief experiment using ChatGPT to extract specific metadata from an open-source scientific journal publication and map it to BioPortal classes. In Part 2, we will share the results of our tests with OntoGPT. To conduct larger biomedical studies, it’s essential to integrate and harmonize datasets from various sources. This requires annotating data using consistent terminology or ontology classes. This standardization is crucial for machine learning and AI research, which rely on standardized datasets.
OntoGPT for the Bioinformatician with Bash Shell Skills
Similar to the previous goal, OntoGPT uses OpenAI to extract annotation from scientific publications using specific ontologies that are valued by researchers, such as GO, CHEBI, MESH, NCIT and others. The tests described below were performed with OntoGPT version 0.3.15. Implementing this solution requires basic coding skills when using existing templates. When custom templates are needed, there is a learning curve involved in understanding a temple’s structure and design options.
Using Existing Templates
OntoGPT provides templates for several types of annotations especially geared towards extracting gene-related information, but also treatment, drugs and diseases. The templates consist of pre-defined LinkML data models described in the next section. With these templates, it is easier to extract ontology annotations from specific text in a particular format. The main task for a bioinformatician, which requires some troubleshooting, is the installation of OntoGPT. OntoGPT requires Python 3.9 or higher and requires installation of OpenAI. We settled for Python 3.10 as later versions gave us errors. With OpenAI one may encounter errors with deprecated functions, such as OpenAI.Completion which has changed. OpenAI has a feature to migrate code to the newer supported code with ‘openai migrate’ but depending on the Python version it may fail. This last error can be solved by upgrading openai. Once installed a bioinformatician can, for example, extract information from text using prompts in a bash shell terminal like this:
echo "One treatment for high blood pressure is carvedilol." > example.txt
ontogpt extract -i example.txt -t drugThe result is written to the screen with the raw_completion_output showing drug and disease was identified:
---
raw_completion_output: |-
disease: high blood pressure
drug: carvedilol
mechanism_links: carvedilol treats high blood pressureAt the end of the result one can find the ontology annotations:
extracted_object:
disease: MONDO:0005044
drug: drugbank:DB01136
mechanism_links:
- subject: MESH:C043211
predicate: biolink:treats
object: AUTO:high%20blood%20pressure
named_entities:
- id: MONDO:0005044
label: high blood pressure
- id: drugbank:DB01136
label: carvedilol
- id: MESH:C043211
label: carvedilol
- id: biolink:treats
label: treats
- id: AUTO:high%20blood%20pressure
label: high blood pressure
Using Custom Schemas
For use cases that require custom metadata annotation, OntoGPT has a guide on developing Custom Schemas. When studying the schema structure, it is important to understand that OntoGPT uses SPIRES (Structured Prompt Interrogation and Recursive Extraction of Semantics) for knowledge graph engineering using OpenAI.
SPIRES allows for grounding of identified entities and concepts using ontology-lookup (via OAK), and dynamic value sets. The process extracts nested semantic structures guided by a schema. It then maps the extraction against existing knowledge bases, a process known as “grounding”. SPIRES uses the LinkML schema system, a YAML-based language for describing ontologies. It automatically compiles the YAML schemas into Pydantic models, which are subsequently used to construct prompts for GPT completion. Therefore, the only thing needed is to create the YAML file following the LinkML system as described in the Custom Schemas guide. The only other article describing the use of ontogpt was found here.
Below is an excerpt of a template created for FAIRLYZ:
classes:
Study:
tree_root: true
attributes:
studysystemmodels:
description: the organism studied,
and when studying microorganisms or cell culture, then the host organism,
also the cultured microorganism if no host is mentioned
range: StudySystemModel
multivalued: true
annotations:
prompt.example: human, mouse, monkey, zebrafish, candida albicans
studytypes:
description: the type of biomedical or clinical study
range: StudyType
multivalued: true
annotations:
prompt.example: Clinical Trial, Clinical Study, Observational Study,
Laboratory Study, Microbiology, Animal Study, AI/ML,
Meta-Analysis, Bioinformatics
…
StudySystemModel:
is_a: NamedEntity
id_prefixes:
- NCIT
annotations:
annotators: bioportal:ncit
StudyType:
is_a: NamedEntity
id_prefixes:
- NCIT
- MESH
- EDAM
annotations:
annotators: bioportal:ncit, bioportal:mesh, bioportal:edam
…
You may then use the schema like any other. For example, if your schema is named fairlyz-study.yaml, then an extract command is:
ontogpt extract -t fairlyz-study.yaml -i input.txtRunning this (or any other command including your custom schema) will install it for future use with OntoGPT, so in subsequent commands it can be referred to by its name (e.g., fairlyz, without the file extension or a full filepath).
ontogpt extract -t fairlyz -i example.txt --output example.output.fairlyz.ymlTo run ontogpt on a PubMed paper or a collection of PubMed IDs run ontogpt pubmed-annotate. We ran it on the same publication as when using the ChatGPT-4 chat prompt earlier:
ontogpt pubmed-annotate -t fairlyz_study.yaml "38476940"Results show that chatgpt completion extracted these terms:
raw_completion_output: |-
raw_completion_output: |-
studysystemmodels: <Humans, SARS-CoV-2, COVID-19; Candida albicans; Staphylococcus aureus>
studytypes: <cross-sectional study>
studydataused: <DNA metagenomics; RNA virome sequencing; medical records>
datasources: <saliva>
studyfocuses: <COVID-19; secondary bacterial infections; sepsis; antimicrobial stewardship; salivary microbiome> OntoGPT then mapped them to these ontologies but failed to map to NCIT classes using bioportal:ncit:
extracted_object:
studysystemmodels:
- <Humans, SARS-CoV-2, COVID-19
- Candida albicans
- Staphylococcus aureus>
studytypes:
- <cross-sectional study>
studydataused:
- EDAM:topic_0654
- EDAM:format_1213
- MESH:D008499
datasources:
- MESH:D012463
studyfocuses:
- MESH:D000086382
- MESH:Q000556
- HP:0100806
- EDAM:topic_3301
- EDAM:topic_3697
named_entities:
- id: EDAM:topic_0654
label: <DNA metagenomics
- id: EDAM:format_1213
label: RNA virome sequencing
- id: MESH:D008499
label: medical records>
- id: MESH:D012463
label: <saliva>
- id: MESH:D000086382
label: <COVID-19
- id: MESH:Q000556
label: secondary bacterial infections
- id: HP:0100806
label: sepsis
- id: EDAM:topic_3301
label: antimicrobial stewardship
- id: EDAM:topic_3697
label: salivary microbiome>
BioPortal’s reliability is crucial for ontoGPT’s performance. During testing, we encountered an error (“ERROR:root:Cannot find slot for studySystemModels…”) due to BioPortal’s downtime, highlighting the importance of verifying the accessibility of ontology websites before executing ontoGPT. This incident emphasizes the need for a preliminary check to ensure uninterrupted ontology access and prevent potential errors.
Summary
We were impressed with the terms ontoGPT extracted from the paper which were accurately categorized but we had to research why some terms were not correctly mapped to one of the selected BioPortal ontologies. The Bioportal version of NCIT is a bit inconsistent about the prefixes it uses. It was recommended to use sqlite:obo:ncit as the annotator instead of bioportal:ncit. This will require writing additional code to identify the BioPortal URI from the sqlite:obo:ncit ID. Overall, we were able to map about 50% of the terms and expect to map more with some fine-tuning.